
|
Sickle Cell Beta Thalassemia Disease
What is Sickle Cell Beta Thalassemia Disease?
Sickle cell beta thalassemia (Hb S/ß Th) is an inherited form of sickle cell disease that affects red blood cells. The most common types of sickle cell disease are SS, SC and S beta thalassemia. Hemoglobin S beta thalassemia disease is the most common sickle cell syndrome seen in people of Mediterranean descent. However, it is also common in people whose families are from Africa, South or Central America, the Caribbean Islands and the Middle East.
A person with sickle cell beta thalassemia disease has red blood cells that sickle due to the hemoglobin S gene, and other red blood cells that are small, pale and misshapen due to the beta thalassemia gene. The severity of the disease varies because the beta thalassemia gene may still produce a small amount of normal hemoglobin. If the gene produces a small amount of normal hemoglobin, b+ thalassemia, individuals have milder symptoms. However, if the beta thalassemia gene produces no normal hemoglobin, b0 thalassemia, the condition is identical to sickle cell disease. Individuals with sickle cell beta thalassemia may have medical problems that include anemia (low hemoglobin), crises (painful episodes), organ damage, infections, lung problems, leg ulcers, bone damage and strokes. Sickle cell beta thalassemia disease is a lifelong condition and requires lifelong treatment.
What is Hemoglobin?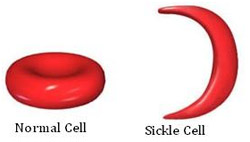
Hemoglobin is found in all red blood cells and carries oxygen from the lungs to tissues and organs throughout the body. Normal red blood cells are soft, smooth, round and can move easily through the body. When affected by sickle cell disease, the red blood cells become rigid, sticky, and sickle shaped. This results in periodic plugging of blood vessels, thereby preventing delivery of oxygen to tissues and organs.
Why is it Important to Know if You Carry Abnormal Hemoglobin?
Like height and eye color, a person inherits genes that produce hemoglobin from each parent. Individuals with sickle cell beta thalassemia disease inherit a hemoglobin S gene from one parent and a beta thalassemia gene from the other parent. It is important to identify people who are carriers of abnormal hemoglobin so they will be aware of their risk of having children with sickle cell disease. If both parents are carriers of sickle cell trait or another hemoglobin change (like beta thalassemia), there is a 25 percent (or one in four) chance that they will have a child with sickle cell disease. 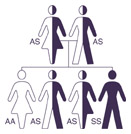 The couple also has a 25 percent chance of having a child with regular hemoglobin (AA) and a 50 percent chance of having a child with a hemoglobin trait like the parents. It is recommended that people who carry a hemoglobin trait meet with a genetic counselor to obtain more information. The couple also has a 25 percent chance of having a child with regular hemoglobin (AA) and a 50 percent chance of having a child with a hemoglobin trait like the parents. It is recommended that people who carry a hemoglobin trait meet with a genetic counselor to obtain more information.
For Additional Information
Illinois Department of Public Health
Genetics/Newborn Screening Program
535 W. Jefferson St., Second Floor
Springfield, IL 62761
217-785-8101
http://www.idph.state.il.us/HealthWellness/genetics.htm

 |
|
Illinois Department
of Public Health
535 West Jefferson Street
Springfield, Illinois 62761
Phone 217-782-4977
Fax 217-782-3987
TTY 800-547-0466
Questions or Comments |
|
